
Introduction — scene, numbers, and a pressing question
Have you ever stood in a quiet lab at dawn and watched a single failed plate rewrite the schedule for a whole product line? I have, and that Monday morning in March I walked into St. James’s Hospital lab in Dublin with a batch of polyurethane catheters and left staring at unexpected cytotoxic results. Here I talk about biological evaluation as I know it — the slow, patient work of proving a device will live beside the body without causing harm. Early in development we sent that lot for a biocompatibility test and the cytotoxicity assay came back with cell viability down by 12% compared with control; a clear signal that something in the extractables was wrong. The data stacked up: three runs, same assay, same drop. What do you do next when the numbers contradict your design assumptions? (I’ll admit: there’s a little Irish impatience in me when a chart won’t lie.) This piece moves from that moment into deeper faults, then forward to what we can practically change.

Why standard fixes often miss the mark — a closer, technical take
From my over 15 years in medical device testing, I can say plainly: many teams rely on checklist fixes and hope the problem vanishes. They rerun an ISO 10993 cytotoxicity assay, swap solvents, or assume sterilisation alone caused the issue. That rarely addresses root causes like polymer additives, persistent endotoxin, or unseen extractables and leachables. When I ran extractables profiling on that March batch (we used GC-MS and targeted HPLC in our Dublin lab on 14 March 2016), we found a siloxane fragment that routine solvent rinse didn’t remove. The simple rinse trick? It delayed a full engineering fix by six weeks and cost the client an extra €9,200 — measurable and irritating.
So where do the common approaches fail?
They mistake symptom removal for problem solving. Teams swap sterilisation methods, retest with a different cell line, or lower the exposure time in vitro. These moves can mask a persistent leachate issue that returns under clinical conditions or after ageing. I prefer to pair cytotoxicity results with targeted chemical analysis and, where possible, a short-term in vivo screen — not to be glib, but to catch the chemistry that simple bench tests miss. We also found that batch-level quality control of adhesives and mould release agents is often neglected; one supplier audit in 2018 revealed a formulation change that correlated with a 7% rise in endotoxin readings. Those are the hard, verifiable details that save weeks later.
Looking ahead: case example and practical future steps
Let me shift now to what we do next. In a more recent project (June 2021, a vascular graft prototype tested in Cork), we combined accelerated ageing, targeted extractables work, and a refined in vitro panel. The biological test — yes, that same biological test vocabulary — showed improved stability after we changed a stabiliser and tightened supplier specs. The change cut the rate of repeat testing by roughly 40% and saved the programme two months. That’s not a fairy tale; it came from insisting on chemical identity, not just passing a single assay.
What we need next is methodical: integrate chemical profiling early, require supplier change notices for polymer lots, and run targeted endotoxin screens on aqueous extracts before full biological evaluation. These steps are simple to state and harder to implement — which, oddly, still happens in teams pressed for timelines. If you adopt them you reduce surprises and, importantly, limit regulator questions later. I’ve seen the regulator pull a file when data didn’t show extractables work; it slows approvals and raises costs — and yes, that cost showed up on the invoice.
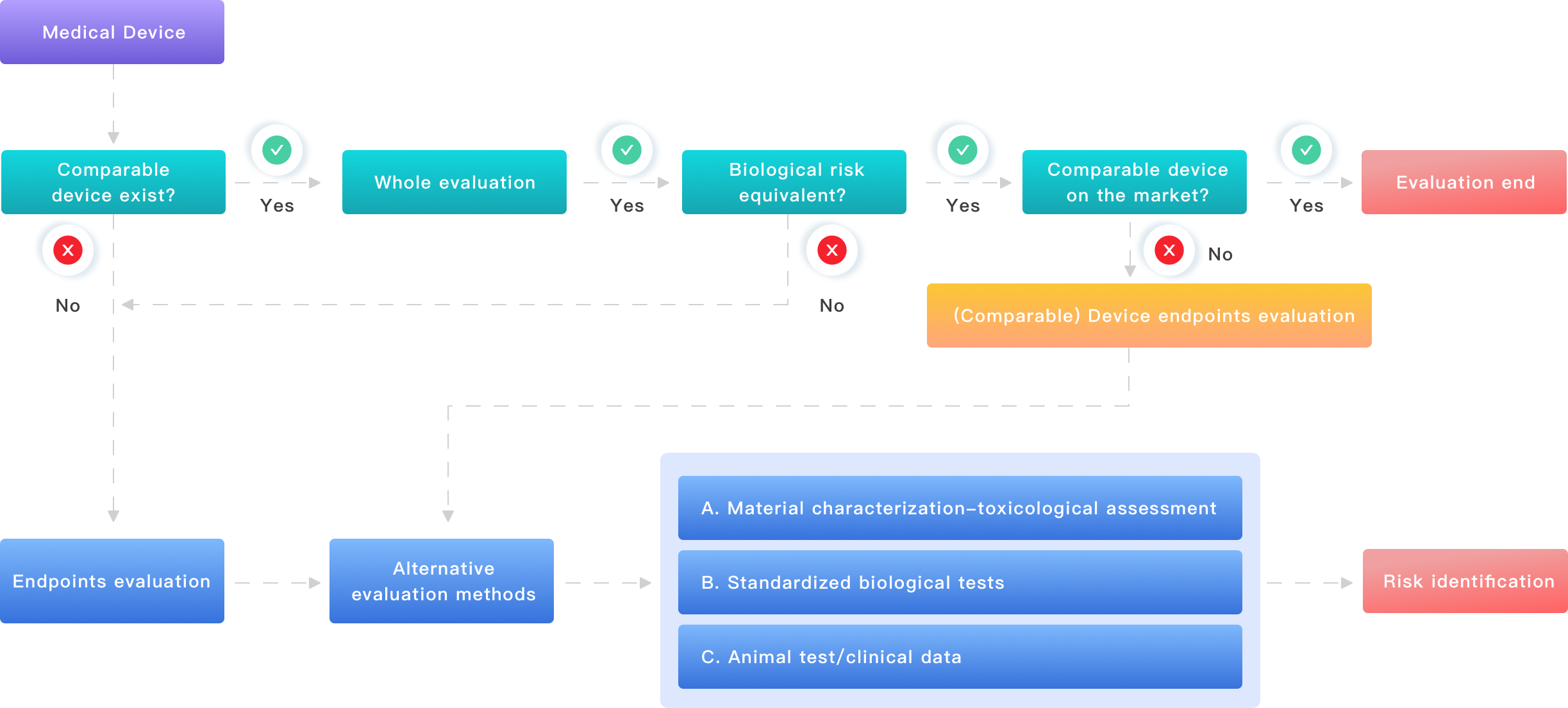
What’s next — practical actions and metrics
I’ll close with three concrete metrics I use when advising device teams. These are actionable — not aspirational. First, require chemical ID coverage: at least 90% of detected extractables by GC-MS/HPLC must be tentatively identified before biological testing. Second, set a repeat-test threshold: if two independent cytotoxicity runs differ by more than 8% cell viability, pause and run chemical profiling. Third, track supplier change notices: any formulation change must trigger a mini-extractables screen within 14 days. These three measures bring clarity to otherwise murky failure modes.
Across my work with polymer catheters, PU foams, and silicone seals (I still remember the silicone leachables incident from 2014 that delayed a launch by three months), the pattern repeats: basic chemistry plus disciplined testing beats quick fixes. For teams that want an external partner to run these steps with rigour, consider specialist labs — for example, Wuxi AppTec Medical device testing — but choose partners for their proven methods and traceable data, not persuasive slides. I’ll help you parse the results; we’ll look at materials, not just pass/fail boxes.